Rett syndrome (RTT) is a rare neurodevelopmental disorder with devastating impact both on patients and their family’s lives, with no cure at present. After a pre- and perinatal period of apparently normal development, RTT patients show stagnation with regression of motor, autonomic and cognitive skills. The disease represents the second most common genetic cause of intellectual disability in girls.
MiND Lab Contribution
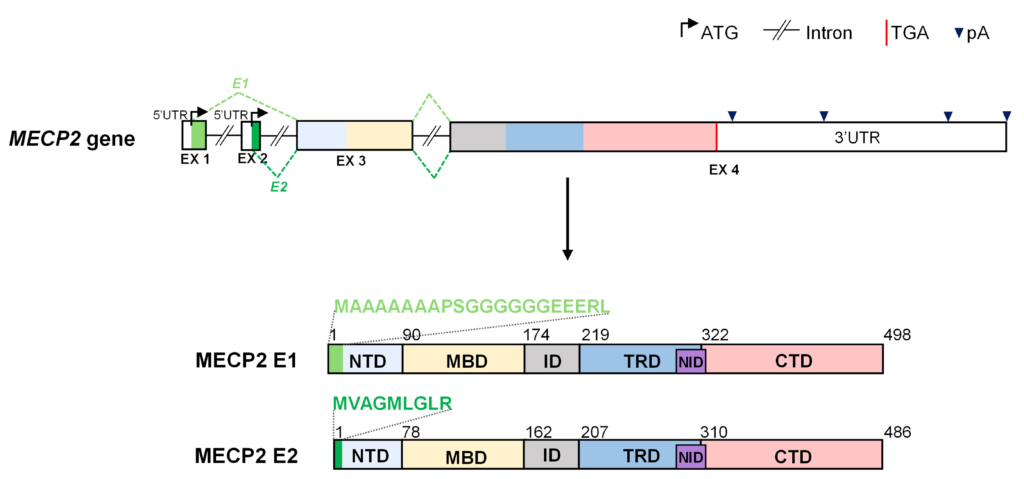
In 1999, MECP2 was identified as the gene that causes Rett syndrome. In 2004, we reported the identification of a new isoform of MECP2, referred to as MECP2_E1, which, while it is very similar to the earlier discovered E2 isoform, is thought to be the disease-relevant form of the gene because it is the abundant isoform expressed in the brain1. This study also reported the first ever RTT patient identified with a disease-causing mutation in exon 1 – an 11bp deletion. We also showed that a single amino acid mutation specific to the E1 isoform, Ala2Val affects the turnover (i.e. degradation) rate of the protein2. No MeCP2 E2-specific RTT-causing mutations have been identified to date.
MeCP2 genetic structure and E1 and E2 protein isoforms domains. EX: exon, UTR: untranslated region, TGA: stop codon, pA: polyadenylation, NTD: N-terminal domain, MBD: methyl CpG DNA binding domain, ID: intervening domain, TRD: transcriptional repression domain, NID: NCoR Interaction Domain, CTD: C-terminal domain. Adapted from Good et al., 2026 (submitted).
In addition to typical Rett Syndrome, MeCP2 mutations have been reported in autism spectrum disorders (ASD), schizophrenia (SCZ), and intellectual disability (ID), and the severity of symptoms can generally be attributed to the location of the mutation on the gene/ protein. To this end, our lab has characterized the functional change caused by mutations that cause disparate phenotypes3, and later identified mutations within an AT-hook in the ID of the protein (which facilitates DNA binding by interacting with AT-rich DNA) that were associated with patients with child onset cognitive decline and schizophrenia4. We further characterized the reduced DNA-binding functionality caused by these mutations. This work exemplifies a core aim of the Vincent lab – to characterize pathogenic neuropsychiatric mutations at the gene and protein levels to expand our knowledge of their cause and to find potential areas of therapeutic targeting to resolve their protein dysfunction.
Current MiND lab research
Several loss-of-function MeCP2 mutations destabilize its protein structure, which not only inhibits its function, but also results in targeted degradation and reduced protein levels. Exogenous expression of a mutant form of MeCP2 in mice alleviates RTT phenotypes5, suggesting that MeCP2 protein abundance is critical for RTT pathology. Treatment of small molecules (SM) targeted to mutations can stabilize proteins in cells3, and a neuroprotective peptide-based therapy is currently under review by the Canadian Drug Agency for treatment of Rett syndrome symptoms5; hence, these molecules show promise for the treatment of patients with destabilizing MeCP2 mutations.
The current MeCP2/ RTT research focus in the MiND lab is to use cellular and in vitro methodologies to identify and functionally validate SMs and peptides that increase the stability (i.e., the abundance) of MeCP2 harboring Rett-syndrome causing mutations as well as ASD, Asperger’s, SCZ, and ID-associated MeCP2 mutations. This work is thus foundational to the Vincent lab’s large undertaking to generate targeted therapies directed towards the numerous pathologies caused by specific MeCP2 mutations.
There are currently no treatments for RTT and other MeCP2-related disorders that target the root cause of the issue (i.e., MeCP2 itself). The MiND lab aims to bridge this gap in available treatments, which could singularly improve the myriad phenotypes caused by mutant MeCP2.
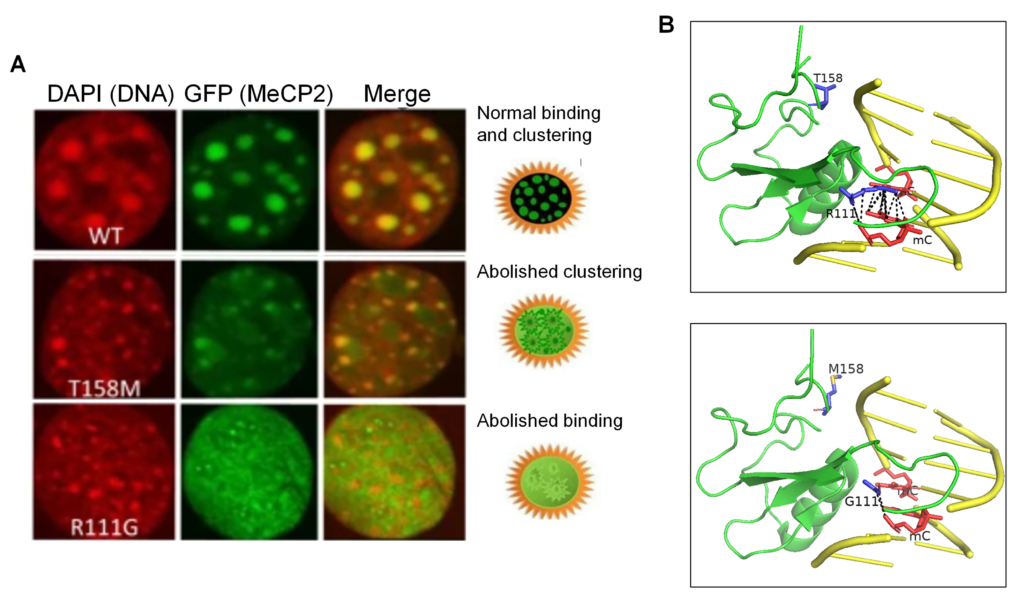
MeCP2 clustering with DNA in cells can be used to assay protein dysfunction. A) Two common RTT-causing mutations under study in the MiND lab show differential clustering in nuclei to the healthy WT protein. Rescue of MeCP2-DNA clustering can be used to test treatment efficacy. Adapted from Sheikh et al., 2016. B) Structural basis for different MeCP2 mutation severities. (Top) R111 makes several contacts with methylated DNA (indicated by dotted lines), whereas T158 is not as strongly associated with DNA, but is important for the overall integrity of the Methyl DNA Binding Domain. (Bottom) At position 111, Glycine (G) is uncharged and smaller than Arginine (R) and is unable to effectively interact with DNA. Methionine (M) is large and hydrophobic compared to Threonine (T), which may impede structure and ability to contact surrounding water molecules. Adapted from PDB: 3C2I. DNA molecule is in yellow and methylated cytosines in red. MeCP2 is in green and residues of interest are blue.